Using RE-EDS to calculate relative binding free energies for ligand scaffold hopping
Calculating the relative binding free energies of a set of ligands to a particular protein is a key application of computational chemistry, facilitating the design of new drug candidates, or explaining experimental results. We have developed the molecular-dynamics (MD) based free energy method RE-EDS [1,2], which is a combination of Hamiltonian Replica Exchange (RE) [3] and Enveloping Distribution Sampling (EDS) [4,5]. RE is a standard approach to enhance sampling in MD simulations. A major advantage of EDS over other free energy methods is the possibility to calculate free energy differences between multiple end states (e.g., multiple ligands) from a single simulation [5]. This feature improves efficiency by up to an order of magnitude compared to pairwise methods. To enable sampling of all end states in a simulation, EDS uses a reference state Hamiltonian.
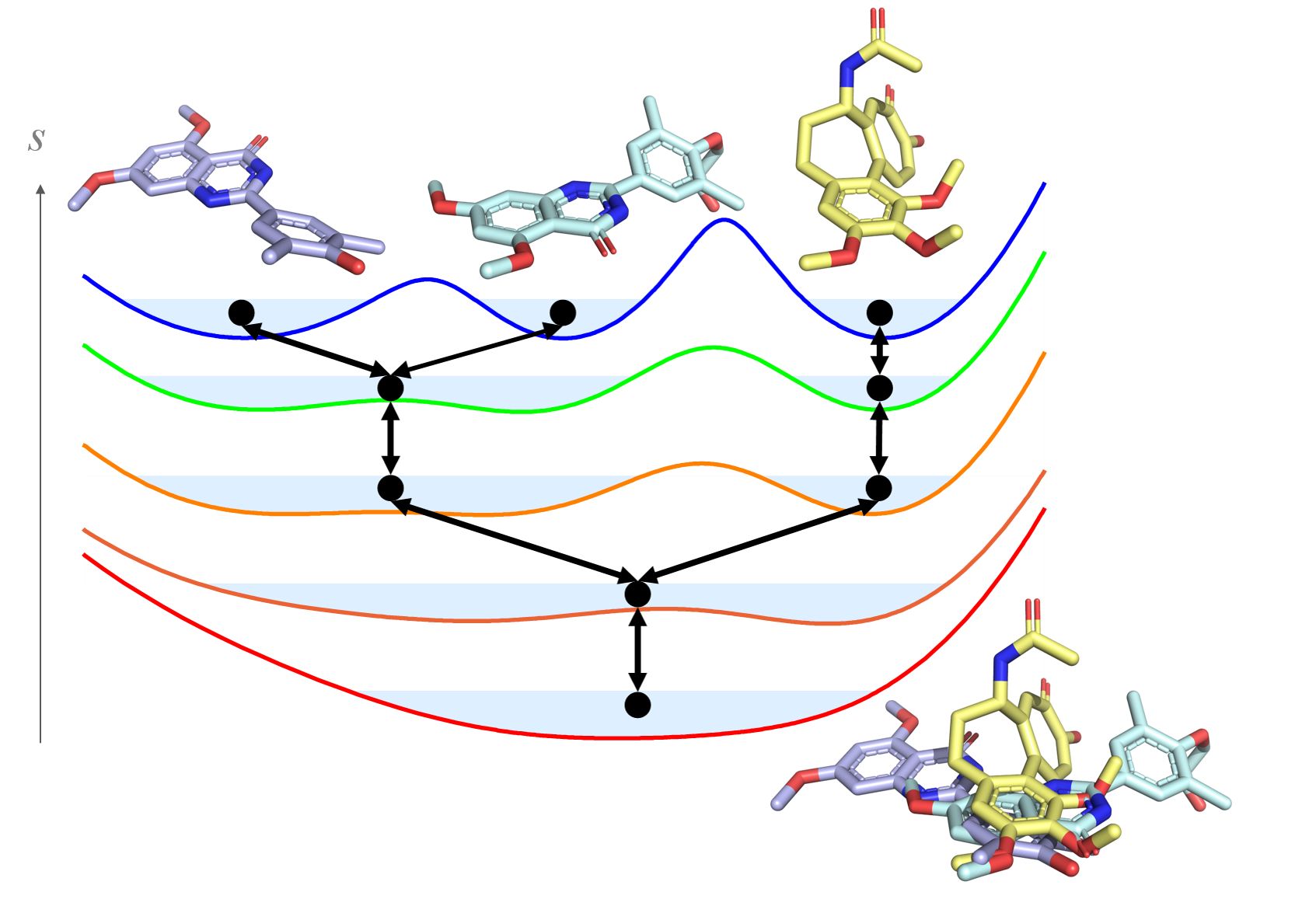
This reference state envelops all ligands in the system and can be modified by two parameters for optimal sampling. The first parameter is the smoothing parameter s, which can lower the energy barriers between the ligands such that transitions from one state to another occur. The second parameter is a vector containing the so-called energy offsets, which are used to align the potential-energy landscapes of all ligands, and, therefore, allow equal sampling of all ligands. However, the determination of the optimal reference-state parameters is challenging and time-consuming for more than two end-states. This issue can be addressed with RE-EDS. In RE-EDS, replicas of the reference state are simulated with different smoothness parameters. At regular time intervals, exchanges of the configurations between replicas are attempted, which enables transitions between end states. This simplifies the choice of the reference state parameters and allows the simulation of ligands with more diverse structures compared to conventional free energy methods. Here, we explore the possible structural diversity of ligands in relative binding free energy calculations with RE-EDS.
[1] D. Sidler, A. Schwaninger and S. Riniker, J. Chem. Phys., 145(15), 154114, 2016
[2] D. Sidler, M. Cristòfol-Clough, and S. Riniker, J. Chem. Theory Comput., 13, 3020–3030, 2017
[3] Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 314, 141–151, 1999
[4] C. D. Christ and W. F. van Gunsteren, J. Chem. Phys., 126, 18, 184110, 2007
[5] C. D. Christ and W. F. van Gunsteren, J. Chem. Phys., 128, 174112,2008