Alchemical Perturbation Density Functional Theory (APDFT)
We introduce an orbital free electron density functional approximation based on alchemical perturbation theory. Given convergent perturbations of a suitable reference system, the accuracy of popular self-consistent Kohn-Sham density functional estimates of properties of new molecules can be systematically surpassed - at negligible cost.
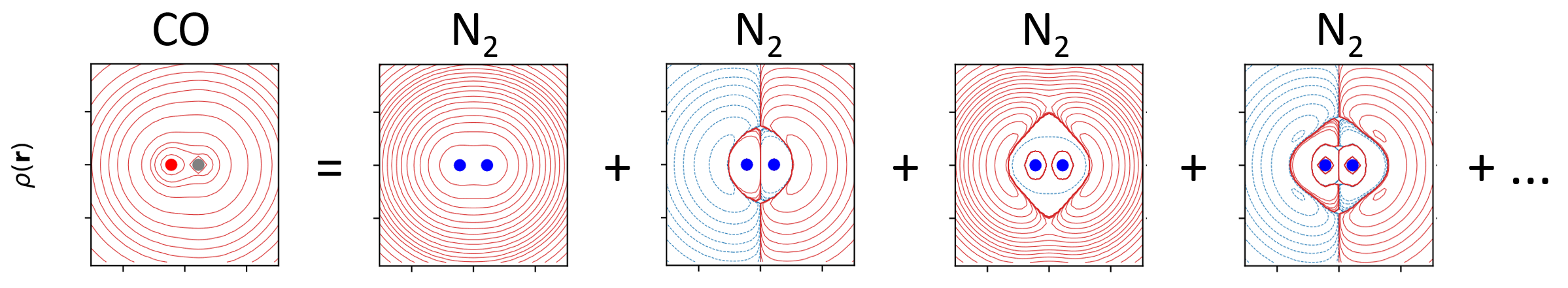
For example, using the CCSD solution for N2, APDFT calculated properties of CO are more accurate than PBE already at 1st order (energies and dipole moments) and 2nd order (quadrupole-moments and forces). The associated energy functional is an approximation to the integrated energy derivative, requiring only perturbed reference electron densities: No self-consistent field equations are necessary to estimate energies and electron densities. Instead, our approach relies on the electron density response w.r.t nuclear charges and treats changes of nuclear charges at any sites as perturbations to the system. We show that the resulting expansion in perturbation orders converges quickly by analytical proof for the hydrogenic atom and for any free atom. Numerical convergence is shown for alchemical perturbations of H2, N2, and benzene.
APDFT based estimates of the electron density of a target molecule are obtained for the same perturbations.
Estimated electronic ground state properties considered include covalent bonding potentials, atomic forces, as well as dipole and quadrupole moments.
APDFT is widely applicable to any level of theory that makes electron densities available and allows to assess a combinatorial number of molecules with one fixed set of calculations rather than calculating molecules one-by-one.
If the perturbation series converges and if the reference level of theory is of sufficient quality, APDFT represents a systematically improving DFT approximation of hierarchies of accuracy.
G. F. von Rudorff, O. A. von Lilienfeld, 2019, arXiv:1809.01647