Learning (from) the electron density: focus on non-covalent interactions and transferability
Intermolecular interactions are the cornerstone of chemistry beyond the single isolated molecule. Their undisputed importance is readily demonstrated by the intensive research effort spent achieving ever more accurate quantification of their magnitude and a chemically more intuitive characterization of their spatial behavior. Among all the molecular properties that encode the relevant information needed to fully characterize both intra- and intermolecular interactions, the molecular charge density occupies a preferential place due to its observable nature and its dependence only on three spatial variables. While, ρ(r) can be accurately obtained solving the electronic structure problem through ab-initio computations, this approach can become rapidly demanding if the electron density has to be evaluated for thousands of different molecules or for the very large chemical systems, such as peptides and proteins. To address this problem, we present a transferable and scalable machine-learning model of the electron density, able to predict the full density field directly from the atomic coordinates.1 Additionally, we show how the regression model can be applied to access the information about intermolecular interactions in a chemically diverse ensemble of molecules derived from the BioFragment database (BFDb).2 Finally, we demonstrate the transferability of the model to new chemical situations, by predicting and analyzing the electron density field of a complex polypeptide.
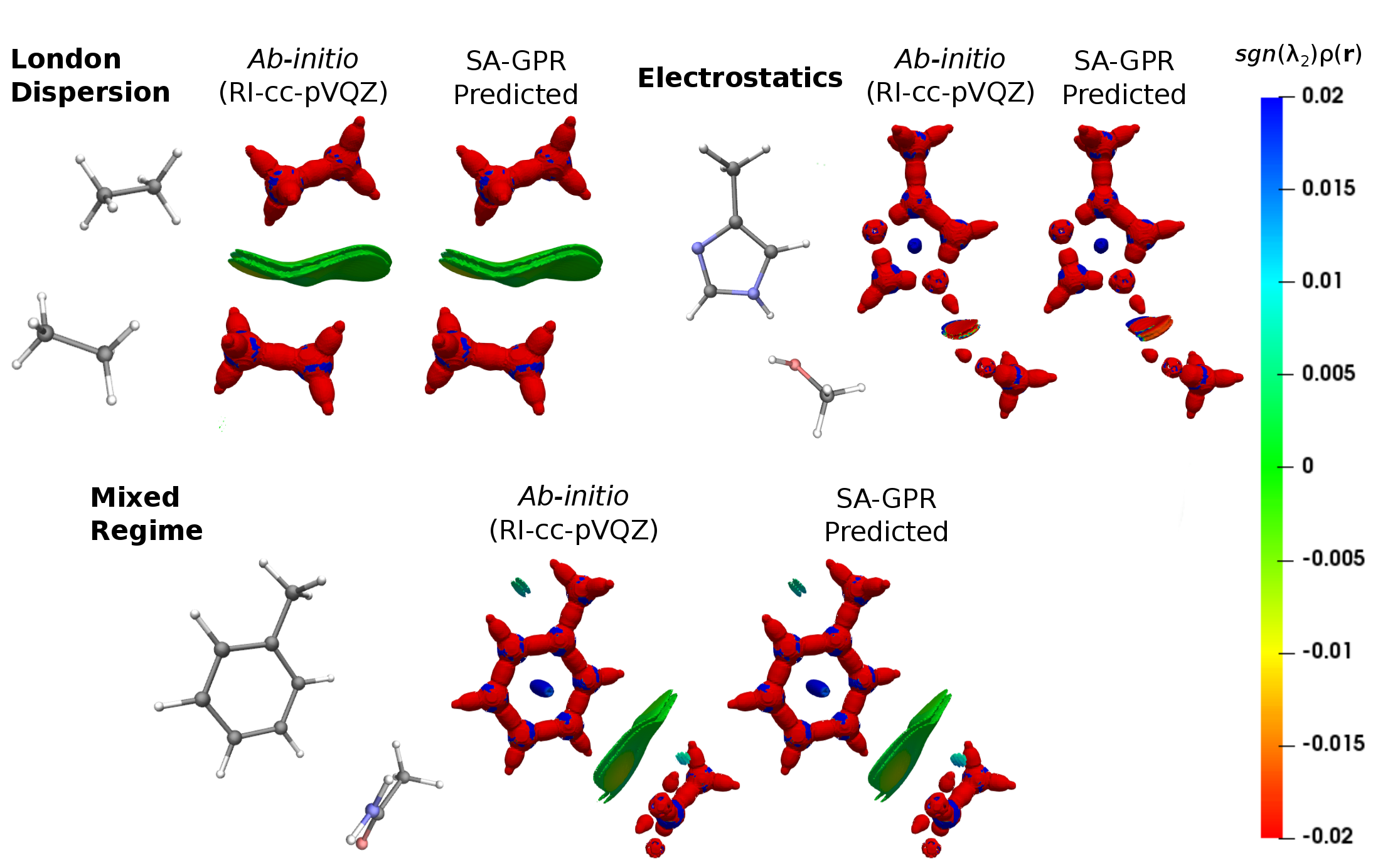
[1] Grisafi, A.; Fabrizio, A.; Meyer, B.; Wilkins, D. M.; Corminboeuf, C.; Ceriotti, M. ACS Cent. Sci. 2019, 5, 57–64.
[2] Burns, L. A.; Faver, J. C.; Zheng, Z.; Marshall, M. S.; Smith, D. G. A.; Vanommeslaeghe, K.; MacKerell, A. D.; Merz, K. M.; Sherrill, C. D. J. Chem. Phys. 2017, 147 (16), 161727.